- Гематологические исследования
- Биохимические исследования
- Биохимическое исследование крови и мочи
- Специфические белки в сыворотке крови и моче
- Биохимические исследования пункционной жидкости
- Биохимические исследования кала
- Биохимические исследования наследственных нарушений обмена веществ
- Исследования при мочекаменной болезни
- Витамины, микроэлементы, оксидативный стресс
- Жирные кислоты
- Фракция трансферрина при алкоголизме
- Неинвазивные маркеры заболеваний печени
- Химический анализ камней
- Эндокринологические маркеры
- Гормоны, участвующие в углеводном обмене
- Гормоны, участвующие в процессе роста
- Гормоны, секретируемые адипоцитами
- Маркеры фертильности
- Маркеры нормальной / патологической беременности
- Гормоны надпочечников
- Гормоны щитовидной железы
- Нейрогормоны
- Пренатальный скрининг на аномалии плода
- Ренин-ангиотензин-альдостероновая система
- Онкологические маркеры
- Маркеры вирусных инфекций
- Маркеры сердечно-сосудистых патологий
- Исследование анемий
- Маркеры патологии костной системы
- Маркеры аутоиммунных болезней
- Антиспермальные антитела
- Аутоантитела при эндокринных, сердечных, почечных заболеваниях
- Аутоантитела при неврологических заболеваниях
- Аутоантитела при дерматологических заболеваниях
- Аутоантитела при пернициозной анемии
- Аутоантитела при сахарном диабете
- Маркеры аутоиммунных заболеваний печени и желудочно-кишечного тракта
- Маркеры ревматических заболеваний и васкулитов
- Маркеры для наблюдения за развитием и лечением болезней
- Маркеры антифосфолипидного синдрома
- Серологические исследования инфекционных болезней
- Аллергологические и иммунологические исследования
- Молекулярно-биологические исследования
- Цитогенетические исследования
- Микробиологические исследования
- Токсикология
- Цервико-вагинальная цитология
- Гистопатологические исследования
- Генетическое консультирование
- Uncategorized

Муковисцидоз – расширенная панель (полное секвенирование CFTR)
 венозная кровь
венозная кровь Общая информация и рекомендации для проведения генетического теста
Кистозный фиброз или муковисцидоз является наиболее распространенным моногенным заболеванием, с частотой 1: 2500-3500 новорожденных среди населения европеоидного происхождения. Он передается аутосомно-рецессивно и характеризуется клиническим плейоморфизмом с хроническим развитием, имеющим среднюю продолжительность выживания 37 лет, возраст, который выше у мужчин, чем у женщин.
| Раса или этническая группа | Частота носителей (гетерозигот) | Скорость обнаружения мутаций |
| Американцы африканского происхождения | 1/65 | 77% |
| Ашкеназские Евреи | 1/25 | 99% |
| Американцы азиатского происхождения | 1/90 | 54% |
| Европейцы | 1/25 | 80% |
| Восточноевропейцы | 1/25 | 75% |
| Румыны | 1/30 | 65% |
| Канадцы французского происхождения | 1/25 | 91% |
| Американцы латиноамериканского происхождения | 1/46 | 81% |
| Северные европейцы | 1/25 | 91% |
| Южные европейцы (итальянцы) | 1/25 | 77%5 |
Заболевание вызвано мутациями в гене, расположенном на длинной руке хромосомы 7 (7q31), который кодирует трансмембранный белок CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), который является частью семейства белков с АТФ-азической активностью и функционирует как канал хлора на уровне апикального полюса мембраны эпителиальных клеток. Кроме того, белок также участвует в регуляции натриевых каналов, вмешивается в транспорт HCO3– через мембраны эпителиальных клеток и может действовать как канал для других белков, таких как глутатион. Недавние протеомные исследования показали, что CFTR взаимодействует со многими внутриклеточными белками, но патофизиологическая значимость этих взаимодействий еще не полностью выяснена 1.
После обнаружения в 1989 году генетического дефекта на уровне гена, участвующего в муковисцидозе, считалось, что ограниченное количество мутаций вызывает это заболевание, но до сих пор было описано более 1500 различных мутаций. Почти все это точечные мутации или небольшие делеции (от 1 до 84 п. н.). Однако важно понимать, что большинство из них редки, и функциональные последствия многих из них трудно понять. Фактически, менее 10 мутаций (показано в таблице 2) встречаются с частотой более 1%, в то время как наиболее распространенная мутация во всем мире, характеризующаяся делецией фенилаланина в положении 508 (ΔF508 – Phe508del) (делеция 3 пар оснований из экзона 10), обнаруживается примерно у 30-80% пациентов с муковисцидозом, в зависимости от пораженной этнической группы. В таблице 1 перечислены 25 мутаций, протестированных в нашей лаборатории для постановки диагноза муковисцидоза или носимого теста1;3.
Мутации гена CFTR могут быть сгруппированы в 6 различных классов, разделенных по отношению к их функциональным последствиям на клеточном уровне (рис. 1):
- класс I: белок не синтезируется;
- класс II: CFTR недостаточно обработан на уровне аппарата Гольджи;
- класс III: белок не работает;
- класс IV: CFTR показывает аномальную проводимость;
- класс V: CFTR показывает частичный дефект синтеза;
- класс VI: CFTR ускоренно деградирован1.
Мутации в классах I, II, III более распространены и связаны с недостаточностью поджелудочной железы, тогда как мутации в классах IV, V и VI встречаются реже, а у пациентов нет проявлений поджелудочной железы1.

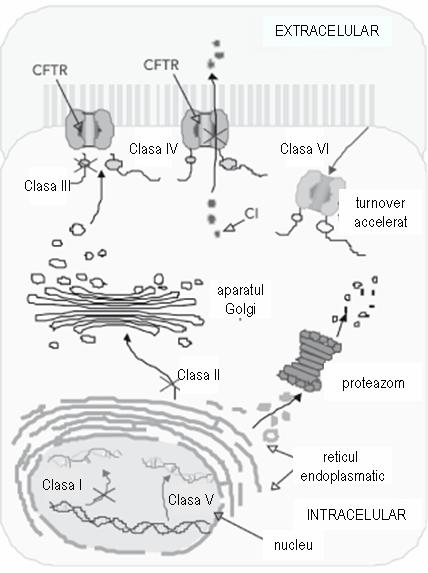
Рисунок 1 адаптиция Роберта Родригеса; Кармен С. Габетта, Карла П. Педро, Фабио Вальдетаро, Мария И. М. Фернандес, Патрисия К. Р. Магальяйнс, Хосе Н.Януарио, Леа М. З. Масиели, кистозный фиброз и скрининг новорожденных, Cad. Общественное здравоохранение vol.24 suppl.4, 2008.
Таблица 1
| 3120+1G>A | A455E | G85E | R334W | 1717-1G>A |
| 3659delC | ΔF508 | R347P | 1898+1G>A | 3849+10kbC>T |
| ΔI507 | N1303K | R553X | 2184delA | 621+1G>T |
| G542X | R1162X | R560T | 2789+5G>A | 711+1G>T |
| G551D | R117H | W1282X | 1078delT | I148T6 |
Таблица 2
| Мутации | Частота | Класс | Фенотип |
| ΔF508 | 66.0% | II | Классический |
| G542X | 2.4% | И | |
| G551D | 1.6% | III | |
| N1303Lys | 1.3% | II | |
| W1282X | 1.2% | И | |
| R553X | 0.7% | И | |
| 621+1G>T | 0.7% | И | |
| 1717-1G>A | 0.6% | И | |
| R117H | 0.3% | IV | Не классический |
| R1162X | 0.3% | Не определенная * | Классическая6 |
* Транскрипция выполняется в нормальных условиях, но белок усечен, вероятно, неправильно упакован, поэтому, скорее всего, он является частью класса II.
Ген CFTR содержит около 250-280 килобаз, состоящих из 27 экзонов. Он кодирует гликопротеин, состоящий из 1480 аминокислот, который образует пять доменов: 2 трансмембранных домена, каждый с 6 отверстиями в α-спирали, 2 нуклеотидных домена (NBD) в цитоплазме, связанных с трансмембранными областями, и регуляторный домен (R), который связывает все трансмембранные Домены. Ионный канал открывается только тогда, когда регуляторная область фосфорилируется протеинкиназой а (PKA), а нуклеотидный домен связывает АТФ.

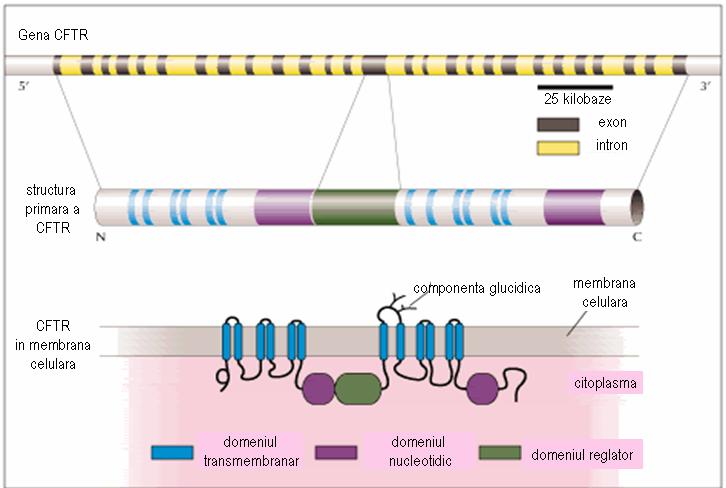
Рисунок 2 Адаптация http://www.chromosome7.htmlplanet.com/custom4.html
Следствием генетических аномалий является отсутствие или неадекватное функционирование хлорных каналов на клеточном уровне, что приводит к изменению транспорта хлоридов в слизистых и серозных железах на уровне большинства органов. Эти выделения будут иметь низкое содержание воды, будут вязкими, прилипшими к эпителию выделительных протоков и трудно выводимыми наружу. Их накопление со временем приводит к изменению функций и разрушению различных органов (легких, поджелудочной железы, печени, кишечника, репродуктивных органов). На коже возникает потовая секреция с высокой концентрацией соли.
Муковисцидоз различается по степени тяжести в зависимости от мутаций CFTR и факторов окружающей среды и проявляется во многих формах, некоторые из которых вызывают раннюю смерть детей в результате прогрессирующей обструктивной болезни легких с бронхоэктазией, другие характеризуются недостаточностью поджелудочной железы и прогрессирующей обструктивной болезнью легких В подростковом возрасте с увеличением частоты госпитализации во взрослом возрасте, а другие проявляются рецидивирующими синуситами и бронхитами или бесплодием у молодых мужчин.
Клиническая картина, возраст на момент постановки диагноза, тяжесть симптомов и скорость прогрессирования заболевания в пораженных органах широко варьируют1;2;6.
Легочные проявления.
В утробе матери, при рождении и сразу после этого у детей с муковисцидозом наблюдается нормальная гистология легких (за исключением протоков подслизистых желез в расширенных дыхательных путях). Однако вскоре после этого происходят изменения в легких, которые характеризуются обструкцией периферических дыхательных путей в результате накопления секрета. У пораженных людей впоследствии развивается воспалительный синдром нижних дыхательных путей, а затем хронические эндобронсические инфекции. Поскольку эластаза (NE), высвобождаемая нейтрофилами в месте воспаления, вызывает расщепление рецептора CR1, компонента c3b комплемента и иммуноглобулина G (IgG), бактерии больше не могут быть опсонизированы и уничтожены, что способствует их стойкости. Изменение противоинфекционной защиты приводит к появлению бактериальных бронхитов и бронхиолитов (чаще всего с Staphylococcus aureus и Pseudomonas aeruginosa, а также Aspergillus fumigatus с развитием мицетом) с интенсивной нейтрофильной инфильтрацией и обструкцией дыхательных путей. Присутствие эластазы также вызывает рост в эпителиальных клетках продукции IL-8, который является хемоаттрактантом для нейтрофилов, разрушает эластин и действует как секретагог, способствуя поддержанию воспаления и инфекции и, таким образом, вызывая структурные повреждения и нарушения газообмена 2.
Респираторные симптомы могут начаться в первый месяц жизни с кашля, тахипноэ или хрипов, при этом некоторые дети могут даже испытывать тяжелый респираторный дистресс, связанный с бронхиолитом.
Поскольку носовые полипы возникают у 10-32% пациентов с муковисцидозом, их присутствие является показанием для проведения теста на пот. У большинства детей старше 8 месяцев (90%) рецидивирующий синусит рефрактерен к антибиотикотерапии.
У взрослых проявления муковисцидоза характеризуются хроническим кашлем (сначала сухим, прерывистым, согласующимся с инфекционными эпизодами, позже со временем удлиняется, становится пароксизмальным, с ночными обострениями и особенно утром после пробуждения, затем продуктивным), прерывистым отхаркиванием с увеличением выработки мокроты, связанным с изменением ее цвета, легкой лихорадкой, одышкой при физической нагрузке, пневмонией, рецидивирующим бронхиолитом, ателектазом, гиперреактивностью дыхательных путей с появлением рефрактерной астмы и отсутствие реакции на лечение β-адренергическими агентами, бронхоэктазии, кровохарканье, пневмоторакс и надземное и межреберное втягивание. По мере прогрессирования заболевания легких в результате хронических инфекций возникают важные структурные повреждения дыхательных путей (кисты или абсцессы), сопровождаемые фиброзом соседней паренхимы легкого и возникновением обструкции с гиперинфляцией и газоблокировкой. Гипоксемия первоначально возникает во время сна и физических нагрузок, а гиперкапния развивается поздно и является отрицательным прогностическим элементом. Также возникновение легочной гипертензии и развитие легочного пуповины один раз с прогрессирующим паренхиматозным заболеванием представляет собой неблагоприятный прогностический признак, который связан с выживаемостью около 8 месяцев.
Недавнее исследование показало, что пассивное воздействие сигаретного дыма отрицательно влияет на функцию легких у людей с муковисцидозом.
Несмотря на все терапевтические вмешательства заболевания легких остаются основной причиной заболеваемости и смертности при фиброзе кисты2; 3;5; 6.
Желудочно-кишечные проявления. Мутации в гене CFTR вызывают изменение секреции хлорида и воды на уровне кишечника, что может привести к появлению мекониальной подвздошной кишки при рождении (чаще у гомозигот ΔF508) или синдрому дистальной кишечной непроходимости в более позднем возрасте.
Мекониальная подвздошная кишка возникает у 15-20% новорожденных с диагнозом муковисцидоз, что может быть основным проявлением в неонатальном периоде. Синдром мекониального воздействия определяется накоплением липкой мукофекалоидной массы в терминальной подвздошной кишке и слепой кишке, адгезией к стенке кишечника, которая иногда может кальцифицироваться.
Атрезия тощей кишки, обычно связанная с заворотом и мекониальным перитонитом, также может быть формой проявления муковисцидоза.
Диагноз муковисцидоза может быть заподозрен с внутриутробной жизни из-за ультразвуковых данных, указывающих на кишечную непроходимость, такую как гиперэхогенный аспект кишечника плода.
Причина синдрома дистальной кишечной непроходимости неясна, что может быть связано с обезвоживанием, лихорадкой, снижением количества ферментативных добавок, заболеванием печени или применением препаратов с антиперистальтическим действием (опиатов, холинолитиков). Клинически он характеризуется повторяющимися спазмами, наличием пальпируемой массы в брюшной полости и признаками полной или частичной кишечной непроходимости. 50% этих пациентов страдают такими осложнениями, как перитонит, заворот, атрезия, некроз, перфорация или образование псевдокисты.
Мукоидное воздействие аппендикса можно наблюдать как бессимптомную пальпируемую массу в Нижнем брюшном квадранте.
В результате повышения внутрибрюшного давления, вторичного по отношению к кашлю, у этих пациентов часто развивается выпадение прямой кишки.
Фиброзная колонопатия-это сущность, характеризующаяся стриктурой восходящей толстой кишки, которая была первоначально описана в 1994 году и является характеристикой муковисцидоза. Высокие дозы ферментов поджелудочной железы, особенно липаз, были криминализированы в этиологии заболевания. Начальные симптомы аналогичны симптомам обструктивного синдрома, и диагноз устанавливается после выполнения баритированного транзита.
Заболеваемость желудочно-кишечными опухолями (пищеводными, желудочными, кишечными, печеночными, желчными, панкреатическими и забрюшинными) увеличивается у пациентов с муковисцидозом, причем толстая кишка является наиболее распространенной локализацией рака.
Экзокринная недостаточность поджелудочной железы (вызванная обструкцией протоков поджелудочной железы секретами, появлением ферментативного аутогестирования и, наконец, интерстициального фиброза), сопровождаемая синдромом мальабсорбции, возникает с рождения у подавляющего большинства людей с диагнозом ФК, за исключением тех, у кого есть определенные мутации CFTR и не требуется введение ферментов поджелудочной железы. Однако они рискуют развить острый или рецидивирующий панкреатит.
Клиническими проявлениями являются стеаторея, вздутие живота, боль в животе, метеоризм, задержка роста (вызванная синдромом мальабсорбции и гемолитической анемией), дефекты свертывания крови или сыпь, связанные с дефицитом жирорастворимых витаминов (A, D, E и K) и цинк. Этим пациентам рекомендуется введение ферментов поджелудочной железы. Люди с FC и функцией поджелудочной железы nomala имеют более мягкое клиническое развитие с более высокой средней выживаемостью (56 лет) по сравнению с людьми с недостаточностью поджелудочной железы.
У детей дефицит витаминов, электролитов и белков проявляется: выраженными родничками; гемолитической анемией из-за дефицита витамина Е; геморрагическими осложнениями, которые могут возникнуть из-за дефицита витамина К из-за печеночной недостаточности или мальабсорбции; гипопротеинемией и отеками; гепатомегалией, сопровождающейся повышением значений ферментов печени; энтеропатическим акродермитом, запорами, гипонатриемическим/гипохлоремическим обезвоживанием и соленой кожей, гипокалиемическим метаболическим алкалозом, вторичным по отношению к хронической потере соли.
Снижение толерантности к глюкозе часто встречается у больных с муковисцидозом (в среднем 40%). Сахарный диабет, связанный с муковисцидозом (DZAMV), проявляется у подростков (кетоацидозом и кетонурией), диагностируется у 7% пациентов в возрасте от 11 до 17 лет с увеличением распространенности в зрелом возрасте. Это чаще встречается у гомозигот ΔF508 и женского пола. DZAMV представляет собой отдельную сущность от диабета 1 или 2 типа, но имеет общие характеристики с обоими типами. Этиология представляет собой комбинацию сниженной секреции инсулина (вторичной по отношению к фиброзу поджелудочной железы и уменьшенному количеству островковых клеток) и периферической резистентности к инсулину. Возникновение этого типа диабета связано со снижением функции легких, более плохим состоянием питания и более короткой выживаемостью.
С увеличением периода выживания у этих пациентов гепатобилиарные заболевания, связанные с муковисцидозом, стали серьезным и частым осложнением, которое может повлиять на качество жизни пациентов. Гепатобилиарное вовлечение становится клинически очевидным после первых 10 лет жизни.
Отсутствие функционального белка CFTR в эпителиальных клетках, выстилающих желчные протоки, вызывает образование сосудистого секрета, который закупоривает канальцы. Если этот процесс обширен, возникает обструктивный цирроз, который может осложниться варикозным расширением вен пищевода, спленомегалией и гиперпленизмом.
Новорожденные с муковисцидозом могут испытывать длительную обструктивную желтуху через внутрипеченочный и внепеченочный желчный застой.
Камни в желчном пузыре имеют более высокую распространенность у пациентов с муковисцидозом (15% молодых людей с муковисцидозом), чем у нормальных субъектов.
Заболевание печени является второй по значимости причиной смертности после заболеваний легких при кистозном фиброзе 2;3; 5; 6.
Плодородие. Более 95% мужчин с ФК бесплодны в результате двустороннего врожденного отсутствия семявыносящих протоков и реже при обструктивной азооспермии. Тело, хвост придатка яичка и семенные пузырьки могут быть аномально расширены или даже отсутствуют. Только 1% пациентов с муковисцидозом фертильны, у них легкие формы заболевания. Сперматогенез является нормальным у этих мужчин.
Женщины с ФК фертильны, хотя некоторые (20%) могут иметь аномальную цервикальную слизь, которая способствует бесплодию.
Поскольку выживаемость людей с ФК значительно улучшилась за последние несколько десятилетий, беременность у женщин с ФК стала важной проблемой. Есть исследования, в которых говорится, что беременность протекает хорошо у этих пациентов, а также исследования, подтверждающие возникновение осложнений.
Отрицательные прогностические факторы, как для матери, так и для плода, в случае беременности-это сила жизненной силы (CVF) менее 50% от прогнозируемой стоимости и плохое состояние питания. Фактически, значение CVF менее 50% от прогнозируемого значения является абсолютным противопоказанием к беременности.
Тем не менее, адекватное лечение заболеваний легких, агрессивное управление инфекциями с помощью широкого спектра антибиотиков и улучшение питания сделали беременность хорошо переносимой, особенно у женщин с легкими или умеренными формами. "К этим факторам риска ухудшения здоровья и ранней смерти после беременности так же, как и для невесомого взрослого населения". В недавнем исследовании Goss et al рассмотрел значение VEF (принудительный объем выдоха), вес, рост и частоту обострения легких в год и обнаружил, что беременность не связана с повышенным риском смерти. Фактически, беременность, по-видимому, не представляет дополнительных рисков ни для подгрупп женщин с диабетом, ни для ОФВ менее 40% от прогнозируемого значения. Важными прогностическими факторами на протяжении всей беременности являются тяжесть материнской легочной недостаточности и состояние питания в том смысле, что их повреждение может ускорить преждевременные роды. Несмотря на все усилия, уровень живых новорожденных у женщин с муковисцидозом в возрасте от 13 до 45 лет составляет 1.9%.
Риск врожденных аномалий у плода не повышен, а грудное вскармливание не является противопоказанием 2;3; 6.
Остеоартикулярные проявления. Гипертрофическая остеоартропатия поражает более 6% больных, чаще встречается у маленьких детей и чаще встречается у мужчин (2:1). Это синдром, характеризующийся аномальной пролиферацией костной ткани в дистальных конечностях длинных костей с появлением цифрового Гиппократа, боли или обычно симметричной суставной опухоли и периостита.
Артропатия при муковисцидозе возникает у 10% больных, может проявляться в любой эволюционный момент. Этиологически задействованы иммунологические механизмы, вероятно, связанные с хронической инфекцией Pseudomonas aeruginosa. Начало происходит внезапно с болью, опухолью и иногда может ассоциироваться с покровными поражениями, такими как макулопапулярная сыпь, узловатая эритема и иногда пурпура. Это может быть моно-или полиартикуляр, поражающий большие или маленькие суставы, наиболее распространенными из которых являются колено, лодыжка, сустав руки, локтя или плеча.
Остеопения или остеопороз являются частыми проявлениями пациенты с ФК и связаны с появлением переломов и кифозов3.
Классический диагноз муковисцидоза устанавливается на основе характерных клинико-анамнестических элементов и затем подтверждается анализом пота или молекулярным анализом.
У 70% пациентов диагноз ставится до 1 года, обычно в первые месяцы жизни. Однако есть больные, диагноз которых подтверждается только после 10 лет.
Диагноз ФК может быть установлен у подозрительных людей, если они показывают:
1. одна или несколько фенотипических характеристик ФК;
и
2. доказательство аномалии функции CFTR:
-наличие болезнетворных мутаций в гене CFTR;
или
-2 аномальные значения хлоридов пота при пилокарпиновом количественном ионофорезе (> 60 мэкв / л);
или
- специфические значения разницы в потенциале носа.
Потовый тест остается золотым стандартом при диагностике заболевания и оценивает концентрацию ионов хлора и натрия в поту. Нормальные значения электролитов в поте составляют <40 ммоль/л; положительные значения у детей >60 ммоль/л, А у подростков и молодых людей >70 ммоль/л; двусмысленные значения: от 40 до 60 ммоль/л обязательно повторяются и интерпретируются в клиническом контексте. Концентрация хлорида более 60 ммоль / л в поте при двух разных определениях устанавливает диагноз заболевания.
Ложноположительные результаты могут быть связаны с синдромом Херлера, а ложноотрицательные-с острой потерей соли. Если FC подозревается у человека с гипонатриемией и гипохлореемией, тест на пот следует отложить до восстановления электролитный баланс3;6.
В следующих особых ситуациях генетическое тестирование является первоначальным диагностическим тестом:
- внутриутробная диагностика плода высокого риска (в 2002 году 4% вновь диагностированных лиц были идентифицированы с помощью пренатальной диагностики);
- пренатальное тестирование плода с низким риском, но с ультразвуковыми изображениями, указывающими на заболевание;
- скрининг новорожденных (в 2002 году 12.8% вновь диагностированных людей были идентифицированы с помощью скрининга новорожденных);
- тестирование симптоматических младенцев (с мекониальной подвздошной кишкой), которые слишком малы, чтобы производить достаточный объем пота;
- тестирование симптоматического человека, у которого есть родственники с идентифицированными мутациями CFTR6.
Поскольку муковисцидоз передается аутосомно-рецессивно, на момент зачатия каждый брат пораженного человека имеет 25% шанс быть носителем и проявлять заболевание, 50% вероятность быть бессимптомным носителем и 25% вероятность быть непригодным и не пострадать.
Пренатальное тестирование проводится на клетках плода, полученных с помощью биопсии ворсинок хориуса, взятых примерно на 10-12 неделе беременности или амниоцентеза, обычно примерно на 15-18 неделе внутриутробной жизни.
Потовый тест должен проводиться постнатально у всех пациентов, у которых подозревался муковисцидоз.
Генетическое тестирование играет важную роль в выявлении мутаций, имеющих важное значение для детерминизма определенных фенотипов. Лучшая корреляция между генотипом и фенотипом связана с функцией поджелудочной железы. Наиболее распространенные мутации были разделены на две категории: те, которые вызывают недостаточность поджелудочной железы, и те, которые связаны с нормальной функцией поджелудочной железы (называемые „sufficient pancreatic”, PS). Люди без повреждения поджелудочной железы обычно имеют один или два мутантных аллеля типа PS, которые доминируют в отношении фенотипа поджелудочной железы.
Напротив, корреляция генотип-фенотип, как правило, слабая при заболеваниях легких при муковисцидозе. Заболевания легких у людей с идентичными генотипами сильно различаются, правдоподобным объяснением является вмешательство факторов окружающей среды.
Гетерозиготы с мутациями ΔF508 / A455E имеют лучшую функцию легких по сравнению с гомозиготными особями для ΔF508.
Тяжесть заболевания легких у людей с одной или двумя мутациями R117H зависит от длины Поли-Т-области от уровня интрона 8, поэтому, если у больного наблюдается вариант 5T в цис-конфигурации, обычно развивается заболевание легких, а у людей с вариантом 7T или 9T фенотип сильно варьируется, который может простираться от отсутствия легочных проявлений до умеренных форм заболевания.
Поскольку мутации A455E и R117H связаны с нормальной функцией поджелудочной железы, менее тяжелое заболевание легких, наблюдаемое у этих людей, может быть следствием улучшения состояния питания 6.
Отрицательный генетический тест на целевые мутации не может исключить заболевание. Поскольку в настоящее время описано более 1000 мутаций, на рынке существует несколько диагностических наборов, которые могут идентифицировать наиболее распространенные мутации для определенной географической области или группы населения. Для действительно подозрительных случаев можно обратиться к сложным методам генетического анализа ДНК (секвенирования).
Американский колледж медицинской генетики рекомендует проводить скрининг носителей с использованием панели, которая выделяет 23 мутации, в которую включено большинство мутаций EC с частотой более 0.1% среди населения США в целом. Однако список скрининговых мутаций может быть дополнен другими мутациями для улучшения чувствительности обнаружения для определенных этнических групп.
Генетические тесты доступны для скрининга бессимптомных людей, которые хотят выяснить, являются ли они носителями дефектного гена муковисцидоза или нет, и обычно включают предварительное тестирование, а также консультации о возможном влиянии положительных или отрицательных результатов теста. Этот тип генетического анализа позволяет родителям выяснить, имеют ли они повышенный риск рождения ребенка с муковисцидозом.
Скрининг носителей муковисцидоза рекомендуется следующим людям:
- взрослые, имеющие в семье родственников с муковисцидозом;
- партнеры людей с муковисцидозом; если у одного партнера муковисцидоз, а у другого-дефектный ген муковисцидоза, то у ребенка будет 50% шанс развития заболевания;
- пары, которые хотят зачать детей.
Если исследования показывают, что человек является носителем дефектного гена муковисцидоза, также необходимо тестирование партнера. Чтобы у ребенка развилось заболевание, оба родителя должны быть носителями мутантного гена. Если анализ партнера отрицательный, у ребенка минимальные шансы на развитие boala6.
Собранный образец-венозная кровь 4.
Контейнер для сбора-vacutainer, содержащий EDTA в качестве антикоагулянта 4.
Собранное количество - 5 мл sange4.
Причины отторжения образца-использование гепарина в качестве антикоагулянта; коагулированные или гемолизированные образцы4.
Стабильность образца - 7 дней при 2-8ºC4.
Метод
В нашей лаборатории доступен метод тестирования: секвенирование всех экзонов CFTR.
Отчетность и интерпретация результатов
Выявленные мутации будут сообщены в гене CFTR и соответствующем генотипе 4.
Библиография
1. Felix A Ratjen MD PhD FRCP(C). Cystic Fibrosis: Pathogenesis and Future Treatment Strategies. In Respir Care 2009 May;54(5):595-605.
2. Girish D Sharma. Cystic Fibrosis, www.emedicine.medscape.com, Ref Type: Internet Communication.
3. Ioan Popa, Liviu Pop, Zagorca Popa, Casandra Cilt. Ghid de management in mucoviscidoza– (fibroza chistica).
4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
5. Mayo Clinic/Mayo Medical Laboratories. Test Catalog. Cystic Fibrosis Mutation Analysis, 70-Mutation Panel. www.mayomedicallaboratories.com. Ref Type: Internet Communication
6. Samuel M Moskowitz, James F Chmiel, Darci L Sternen, Edith Cheng, and Garry R Cutting.CFTR- Related Disorders. Gene Reviews, 2008. www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.
