- Гематологические исследования
- Биохимические исследования
- Биохимическое исследование крови и мочи
- Специфические белки в сыворотке крови и моче
- Биохимические исследования пункционной жидкости
- Биохимические исследования кала
- Биохимические исследования наследственных нарушений обмена веществ
- Исследования при мочекаменной болезни
- Витамины, микроэлементы, оксидативный стресс
- Жирные кислоты
- Фракция трансферрина при алкоголизме
- Неинвазивные маркеры заболеваний печени
- Химический анализ камней
- Эндокринологические маркеры
- Гормоны, участвующие в углеводном обмене
- Гормоны, участвующие в процессе роста
- Гормоны, секретируемые адипоцитами
- Маркеры фертильности
- Маркеры нормальной / патологической беременности
- Гормоны надпочечников
- Гормоны щитовидной железы
- Нейрогормоны
- Пренатальный скрининг на аномалии плода
- Ренин-ангиотензин-альдостероновая система
- Онкологические маркеры
- Маркеры вирусных инфекций
- Маркеры сердечно-сосудистых патологий
- Исследование анемий
- Маркеры патологии костной системы
- Маркеры аутоиммунных болезней
- Антиспермальные антитела
- Аутоантитела при эндокринных, сердечных, почечных заболеваниях
- Аутоантитела при неврологических заболеваниях
- Аутоантитела при дерматологических заболеваниях
- Аутоантитела при пернициозной анемии
- Аутоантитела при сахарном диабете
- Маркеры аутоиммунных заболеваний печени и желудочно-кишечного тракта
- Маркеры ревматических заболеваний и васкулитов
- Маркеры для наблюдения за развитием и лечением болезней
- Маркеры антифосфолипидного синдрома
- Серологические исследования инфекционных болезней
- Аллергологические и иммунологические исследования
- Молекулярно-биологические исследования
- Цитогенетические исследования
- Микробиологические исследования
- Токсикология
- Цервико-вагинальная цитология
- Гистопатологические исследования
- Генетическое консультирование
- Uncategorized


Общая информация
Галактоземия-это аутосомно-рецессивное заболевание, передающееся через нарушение метаболизма галактозы с появлением осложнений, которые могут поставить под угрозу жизнь новорожденного5.
Галактоза является компонентом лактозы, гликолипидов, гликопротеинов и протеогликанов; его метаболизм достигается с предварительным воздействием на печень и почки. Преобразование галактозы в глюкозу включает следующие этапы (см. Рисунок 1):
- фосфорилирование галактозы с образованием галактозо-1-фосфата, реакция, которая достигается с помощью галактокиназы;
- перенос остатка галактозила на UDP-глюкозу под действием UDP-глюкозо-галактозо-1-фосфат-уридилтрансферазы (GALT);
- взаимосвязь между глюкозой и галактозой происходит на уровне нуклеозид-дифосфатов, включает изменение стереохимической конфигурации 4’-гидроксильной
- группы и катализируется UDP-глюкозо-4-эпимеразой; этот этап способствует регенерации UDP-глюкозы и, таким образом, обеспечивает непрерывность цикла метаболизма галактозы.
Вырабатываемый глюкозо-1-фосфат может быть изомеризован в глюкозо-6-фосфат, который играет важную роль в процессе гликолиза и биосинтеза инозита.
Галактоза может быть преобразована в галактитол под действием альдозредуктазы в присутствии НАДФН (или НАДН), что представляет собой альтернативный путь метаболизма галактозы у пациентов с галактоземией. Он также может окисляться под действием галактозодегидрогеназы, что приводит к образованию галактоновой кислоты и CO21.

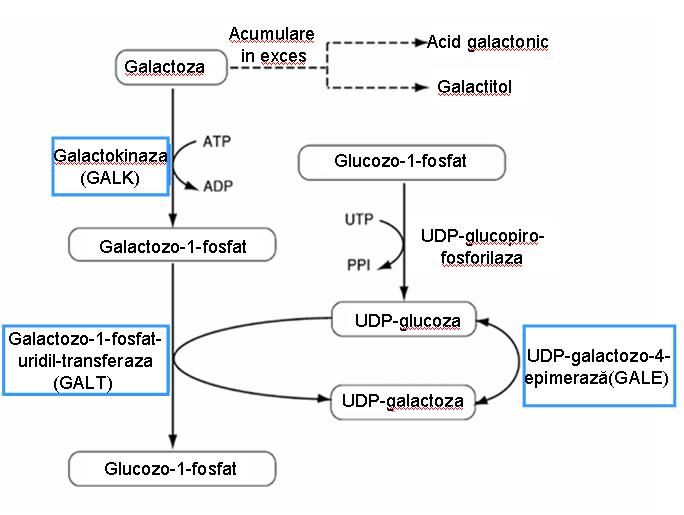
Рис. 1 метаболизм галактозы (путь Лелуара)
Гипергалактоземия связана с 3 ферментативными дефицитами:
– тип I (GALT) – изменение ферментативной активности галактозо-1-фосфат-уридилтрансферазы( GALT), которая отвечает за наследственную галактоземию и является наиболее распространенным ферментативным дефицитом;
-тип II (GALK1) – изменение ферментативной активности галактокиназы, которая превращает галактозу в галактозо-1-фосфат; это редкий дефицит;
-тип III (GALE) – изменение ферментативной активности уридин-дифосфат-глюкозо-4-эпимеразы, которая эпимеризирует UDP-галактозу в UDP-глюкозу, а также является частым3.
| Тип | Ген | Локус | Энзим | Наименование |
| 1 | GALT | 9p13 | галактозо-1-фосфат-уридилтрансфераза | Галактоземия классическая |
| 2 | GALK1 | 17q24 | Галактокиназа | Недостаток галактокиназы |
| 3 | GALE | 1p36-p35 | UDP-галактозо-4-эпимераза | Дефицит UDP-галактозо-4-эпимераза |
Классическая (тип I) форма заболевания и, кстати, самая тяжелая форма определяется мутациями в гене, который кодирует фермент галактозо-1-фосфат-уридилтрансферазу (GALT). Это наиболее распространенное (1/48000 новорожденных) моногенное состояние, которое интересует углеводный обмен. Гомозиготы по классическому аллелю галактоземии (генотип G/G) имеют низкую ферментативную активность ниже 5%, что препятствует эффективному превращению галактозы в глюкозу. Дефицит GALT приводит к накоплению галактозо-1-фосфата и галактозы в тканях, особенно в печени, головном мозге или почках. Считается, что повреждение печени (цирроз) и умственная отсталость, вызванные классической галактоземией, зависят от количества галактозо-1-фосфата, депонированного в этих тканях. Галактоза превращается в галактитол в клетках и оказывает осмотическое действие, такое как отек волокон хрусталика, что может привести к катаракте и нейронам, что может вызвать псевдотумор мозжечка. Как и в форме, связанной с дефицитом галактокиназы, катаракта развивается вторично по отношению к накоплению галактитола в хрусталике. У гомозиготных женщин риск развития гипергонадотропного гипогонадизма с возникновением недостаточности яичников увеличивается и проявляется в раннем возрасте, даже если диетическое лечение назначается незамедлительно. Однако у пострадавших женщин могут быть дети.
Гетерозиготы (генотип G / N, n-нормальный аллель) протекают бессимптомно, что приводит к снижению активности фермента вдвое по сравнению с людьми, у которых нет мутаций 3;5.
Ген, кодирующий фермент GALT, расположен на короткой руке хромосомы 9 и состоит из 11 экзонов (см. Рисунок 2). Было идентифицировано более 180 мутаций гена; в общей сложности 8 болезнетворных мутаций (G) обычно встречаются у пациентов с классической галактоземией. Наиболее распространенной является точечная мутация в экзоне 6, которая вызывает замену глютамина аргинином (Q188R; p.Gln188Arg) и которая отвечает за 60-70% случаев заболевания у кавказцев. Мутация S135L (P.Ser135Leu), вызывающая замену Серина лейцином, чаще всего встречается в популяции черной расы, связанной с более чем 50% случаев заболевания в этой категории людей. Мутация K285N (p.Lys285Asn), вызывающая замену лизина аспарагином в экзоне 9, занимает второе место по частоте в аллелях, связанных с заболеванием, у кавказцев (28%). Мутация L195P (p. Leu195Pro), заменяющая лейцин пролином, регистрируется в 5-7% случаев классической галактоземии 6;7.

Рис. 2 локализация гена GALT на хромосоме 9
(www.wiki.medpedia.com/Galactoso-1-phosphate uridylyltransferase)
В дополнение к классической форме существует Дуарте вариант галактоземии – бессимптомное состояние или сопровождающееся легкими клиническими проявлениями – что является результатом частичного повреждения фермента GALT. Этот вариант характеризуется гетерозиготностью, составленной для классического аллеля G, вызывающего отмеченное снижение GALT, и аллелем Дуарте D-2 (D2; иногда называемым D), вызывающим частичное повреждение GALT. Гемолизаты пациентов с Галт-галактоземией демонстрируют в среднем ферментативную активность ~ 25%, хотя описаны большие индивидуальные вариации. При электрофорезе с изоэлектрическим фокусом достигается характерный аспект измененной подвижности изофермента GALT. Предыдущие исследования показали, что аллели D являются носителями мутации missens p.N314D (c.940A>G), которая вызывает замену аспарагиновой кислоты в положении 314. Фенотипы Дуарте (D/N, D/D и D/G) показывают приблизительно 75, 50 и 25% нормальной активности GALT соответственно и встречаются с распространенностью около 6% среди белой популяции. Замена N314D полностью ответственна за изменение подвижности ферментов, но не объясняет частичное нарушение активности GALT. Кроме того, эта мутация также обнаруживается на аллелях Duarte-1 (D1, также называемых Лос-Анджелесом или Лос-Анджелесом), которые связывают нормальную или повышенную ферментативную активность, что по этой причине считается распространенным вариантом или полиморфизмом гена GALT. Дальнейшие исследования аллелей D1 и D2 показали, что N314D обнаруживается в дисбалансе связи с другими вариантами нуклеотидных последовательностей, и они различаются между D1 и D2.
Таким образом, аллели D1, в частности, демонстрируют изменение нуклеотидов c.652C>t, что вызывает молчаливое замещение на уровне кодона 218 (CTA →TTA, p.L218, также иногда называемое L218L), в то время как аллели D2 показывают делецию промотора 5’ 4 п. н. (c.-119_-116delgtca) вместе с 3 модификациями интронных оснований. Оба аллеля D1 и D2 также несут расширенную последовательность нуклеотидов a на уровне интрона 10. Разница между двумя аллелями в отношении ферментативной активности GALT долгое время оставалась предметом споров. Недавнее исследование указывает на наличие уменьшенной экспрессии мРНК на уровне аллеля D2, что способствует нарушению функции GALT; кроме того, делеция 5’ 4 п. н. промотора будет представлять собой причинную мутацию при галактоземии Duarte2.
Дефицит галактокиназы (тип II) может быть заподозрен у людей с катарактой, галактоземией и повышенной экскрецией галактитола с мочой, но не имеет других клинических проявлений.
Распространенность дефицита Галка неизвестна, но, вероятно, ниже 1:100000. Обнаружение низкого уровня активности галактозокиназы является важным элементом для постановки диагноза, но подтверждение делается путем выявления мутаций в гене, кодирующем фермент.
Он может быть ограничен только эритроцитами и лейкоцитами и связан с развитием катаракты, но не вызывает замедления роста, умственной отсталости или повреждения печени.
Введение лактозы беременным женщинам с дефицитом галактокиназы может вызвать катаракту плода 5.
Дефицит UDP-галактозо-4-эпимеразы (GALE) (тип III) следует подозревать у людей с заболеваниями печени, нейросенсорной глухотой, связанной с повышенными значениями эритроцитарного галактозо-1-фосфата и нормальными уровнями GALT. Диагноз устанавливается на основании обнаружения низкого уровня активности UDP-галактозо-4-эпимеразы и подтверждается путем выявления мутаций в гене, кодирующем фермент. Дефицит UDP-галактозо-4-эпимеразы имеет предполагаемую распространенность 1: 23000 в Японии и неизвестен в других популяциях 3;5.
Клинические проявления классической галактоземии возникают у младенца через несколько дней или недель после рождения и определяются приемом грудного молока или различных молочных смесей, содержащих лактозу. Они состоят из неспособности глотать и рвать, отсутствия роста, гепатоцеллюлярного повреждения (гепатомегалия с печеночной недостаточностью), желтухи, гипогликемии, гиперамониемии, кровотечения и сепсиса. Умственная отсталость становится очевидной через 6-12 месяцев и чаще всего необратима. Новорожденные с классической формой галактоземии восприимчивы к генерализованным бактериальным инфекциям (особенно Escherichia coli), которые могут вызывать Бузину и смерть в неонатальном периоде. Однако, если диета без лактозы установлена на ранней стадии, симптомы быстро исчезают, а осложнения (печеночная недостаточность, сепсис, неонатальная смерть) можно предотвратить. Катаракта обычно не возникает при рождении, но развивается постепенно, будучи видимой в течение нескольких недель или месяцев. Несмотря на соответствующее лечение, у детей с галактоземией в долгосрочной перспективе повышен риск развития задержки роста и умственного развития, проблем с речью (вербальная диспраксия), макронодулярного цирроза печени и недостаточности яичников у женщин. Прогноз обычно сохраняется и варьируется у разных пациентов 3;5; 6.
Галактоземия - это состояние, которое можно обнаружить с помощью скрининга новорожденных. Клинитест является самым простым скрининговым тестом и основан на определении галактозы в образце мочи. Его проводят через 24-36 часов после приема препарата, содержащего лактозу. В некоторых странах используется тест из капли крови, собранной на стандартизированной фильтровальной бумаге (card Guthrie), с помощью которой определяется общая галактоза (галактоза и галактозо-1-фосфат) и/или активность фермента GALT. У детей с высоким риском галактозо-1-фосфат может быть определен из крови, собранной из пуповины. Идеальным скрининговым тестом является тот, который измеряет активность GALT3;5.
Рекомендации по генетическому тестированию
Генетическое тестирование с целью выявления мутаций GALT используется для подтверждения диагноза галактоземии у детей с положительным неонатальным скринингом. Это также полезно для дифференциации аллелей Duarte D2 от аллелей LA.
Таким образом, генетическое тестирование определяет генотип и позволяет установить прогноз.
Тестирование близких родственников на наличие бессимптомного статуса носителя может проводиться в соответствии с генетическим советом и только после выявления болезнетворных мутаций в семье. Поскольку галактоземия передается аутосомно-рецессивно родители ребенка с классической галактоземией обязательно гетерозиготны по мутантному аллелю G, а бессимптомные его братья и сестры имеют риск 2/3 быть носителями аллеля G. также братья и сестры родителей имеют 50-процентный риск быть носителями.
Братья и сестры ребенка с галактоземией Дуарте имеют на момент зачатия 25% риск галактоземии D/G, если у родителей есть генотипы D/N и G/N, и 25% риск классической галактоземии G/G, если родители демонстрируют генотипы D/G и G/N; по этой причине генетическое тестирование родителей оправдано.
Пренатальная диагностика плодов с 25-процентным риском классической галактоземии (г/г) возможна с помощью анализа ДНК, извлеченной из клеток плода, полученных с помощью амниоцентеза, обычно выполняемой примерно на 15-18 неделе беременности или биопсии ворсинок хориуса, примерно на 10-12 неделе беременности. И в этих случаях G-аллели в семьях должны быть идентифицированы заранее. 5.
Собранный образец-венозная кровь 4.
Контейнер для сбора-vacutainer, содержащий EDTA в качестве антикоагулянта 4.
Собранное количество - 5 мл sange4.
Причины отторжения образца-использование гепарина в качестве антикоагулянта; коагулированные или гемолизированные образцы4.
Стабильность образца - 7 дней при 2-8ºC4.
Метод-полное секвенирование гена GALT + анализ делетий / дупликаций MLPA; таким образом, могут быть обнаружены как мутации, связанные с классической галактоземией, так и варианты Duarte1.
Отчетность и интерпретация результатов
Мутации, обнаруженные в гене GALT и соответствующем генотипе, будут сообщены 4.
Библиография
1. David L. Nelson, Michael M. Cox. Glycolysis, Gluconeogenesis, and the Pentose Phosphate Pathway. In Lehninger Principles of Biochemistry, fifth edition, 2008, W. H. Freeman and Company, 545-546.
2. Amanda E. Carney, Rebecca D. Sanders, Kerry R. Garza, Lee Ann McGaha, Lora J. H. Bean, Bradford W. Coffee, James W. Thomas, David J. Cutler, Natalie L. Kurtkaya, Judith L. Fridovich-Keil. Origins, distribution and expression of the Duarte-2 (D2) allele of galactose-1-phosphate uridylyltransferase. In Human Molecular Genetics, 2009, Vol. 18, No.9, 1624-1632.
3. Gerard T Berry, George A Anadiotis, Galactose-1-Phosphate Uridyltransferase Deficiency (Galactosemia) www.emedicine.medscape.com, Ref Type: Internet Communication.
4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
5. Louis J Elsas II. Galactosemia. Gene Reviews, 2000, www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.
6. Mayo Clinic, Mayo Medical Laboratories. Reference Laboratory Services for Health Care Organizations. Galactosemia Gene Analysis, Known Mutation. www.mayomedicallaboratories.com. 2010. Ref Type: Internet Communication.
7. OMIM (Online Mendelian Inheritance in Man). Galactose-1-Phosphate Urydylyltransferase. http://www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.
8. Valeriu Popescu, Alis Antrasian, Andrei Zamfirescu. Screeningul neonatal in bolile genetice de metabolism. In Revista Romana de Pediatrie, volum LVIII, nr.4, 2009.
