- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Uncategorized


Informaţii generale
Coreea Huntington este o afecţiune neurodegenerativa progresiva, cu transmitere autozomal dominanta, caracterizata fenotipic prin miscari involuntare, distonie, declin cognitiv si tulburari comportamentale.
Prevalența bolii este 5-10:100000 cazuri. In mod tipic, primele simptome apar intre 35 si 50 ani insa afecţiunea poate debuta la orice varsta. Din cauza apariţiei tardive a simptomelor, o persoana afectata de coreea Huntington poate avea copii inainte de a sti ca sufera de aceasta boala1;6.
Boala a fost descrisa pentru prima data in 1872 de catre medicul american George Huntington. Termenul „coree” isi are originea in cuvantul grec khoreia = dans si caracterizeaza miscarile involuntare rapide, nerepetitive, ample si bruste specifice bolii; aceste miscari pot varia in severitate, de la agitatie insotita de o exagerare intermitenta usoara a gesturilor si expresiei, miscari nervoase ale mainilor, mers ondulant instabil pana la un flux continuu de miscari violente, invalidante. Coreea este prezenta la peste 90% dintre pacienti si se agraveaza in cursul primilor 10 ani de evolutie. Pe masura ce boala avanseaza coreea este inlocuita treptat de distonie si manifestari parkinsoniene, cum ar fi bradikinezia, rigiditatea, instabilitatea posturala. In stadiile tardive se instaleaza un sindrom akinetic-rigid, in care miscarile coreice sunt minime sau absente. Afectarea functiei motorii voluntare reprezinta un semn precoce de boala; dizartria si tulburarile oculomotorii se dezvolta de asemenea in stadiile initiale, in timp ce disfagia apare tarziu.
Boala Huntigton juvenila (varianta Westfal; 5-10% din cazuri) debuteaza inainte de varsta de 20 ani si se caracterizeaza prin manifestari parkinsoniene, distonie, dementa, epilepsie si coree usoara sau chiar absenta2;5.
Toate persoanele afectate de coreea Huntington prezinta un declin global progresiv al functiilor cognitive. Sindromul de dementa include in stadiile initiale iritabilitate, neglijenta, pierderea interesului; incetinirea procesului de gandire si tulburarile de memorie apar mai tarziu. Acest tablou clinic este caracteristic dementei subcorticale (diferita de cea care apare in boala Alzheimer), despre care se crede ca reflecta disfunctia circuitului neuronal fronto-subcortical.
Tulburarile comportamentale sunt reprezentate in principal de boala afectiva. Depresia este mai prevalenta; doar un numar mic de pacienti prezinta atacuri maniacale caracteristice bolii bipolare. Se poate inregistra o rata mai mare de suicid; persoanele afectate pot dezvolta de asemenea psihoza, simptome obsesiv-compulsive, tulburari sexuale, afectarea somnului si modificari de personalitate2;6.
Trasaturile neuropatologice ale coreei Huntington includ degenerescenta neuronala selectiva din caudat si putamen. Afectarea preferentiala a neuronilor spinosi mijlocii GABA-ergici ai caii indirecte de control al miscarilor din ganglionii bazali constituie baza neurobiologica a coreei (deteriorarea sistemelor colinergice si GABA-ergice din corpii striati afecteaza inhibitia GABA-ergica a celulelor din globus pallidus); interneuronii striatali nu sunt in general afectati; alte regiuni cerebrale ce pot fi implicate sunt: substanta neagra, hipocampul, precum si alte zone din cortex. Au fost propuse mai multe mecanisme pentru distrugerea neuronala, cum ar fi exotoxicitatea (efectul neurotoxic al aminoacizilor excitatori in prezenta activarii excesive a receptorilor postsinaptici), stres-ul oxidativ, perturbarea metabolismului energetic si apoptoza2;5.
HTT (HD) este singura gena asociata cu boala Huntington; unica mutatie descrisa consta in expansiunea repetitiilor trinucleotidului CAG; Nancy Wexler, avand experienta bolii in propria familie, a descoperit in 1983 localizarea genei HD pe bratul scurt al cromozomului 4 (4p16.3).
Gena HTT (numita si IT15) are o lungime de aproximativ 200 kb, cu 67 exoni si o regiune codanta de 10366 bp. La nivelul primului exon se gasesc in alelele normale 8-26 triplete CAG, in timp ce la indivizii afectati de coreea Huntington numarul repetitiilor depaseste 36 (numarul maxim cunoscut pana acum fiind de 250), ceea ce duce la o „alunecare” a ADN-polimerazei in timpul replicarii3;5. Coreea Huntington juvenila se manifesta atunci cand numarul de repetitii depaseste 60. In cazul in care defectul este transmis de la tata, numarul tripletelor este mai mare decat in cazul transmiterii de la mama (datorita fenomenului de amprentare). Sunt descrise 2 tipuri de alelele cauzatoare de boala:
• cu penetranta completa (sunt asociate intotdeauna cu boala), care prezinta > 40 triplete CAG;
• cu penetranta redusa, caracterizate prin 36-39 repetitii CAG, care confera un risc crescut de boala Huntington, dar nu toti purtatorii devin simptomatici.
Alaturi de alele cauzatoare de boala mai exista si alele intermediare (alele „mutabile”), cu 27-35 repetitii CAG, care nu se asociaza cu riscul de a dezvolta simptome de boala, dar care datorita instabilitatii sirului repetitiv CAG, transmis de la o generatie la alta, sunt predispuse la expansiune si pot induce un risc crescut de a da nastere unui copil cu alela cauzatoare de boala. Acesti copii sunt considerati ca avand o mutatie “de novo”6.
Gena HTT codifica o proteina – huntingtina; exista numeroase polimorfisme ale genei care conduc la un numar variat de reziduuri de glutamina (aminoacid codificat de tripletul CAG) ce sunt aliniate monoton la capatul aminoterminal al proteinei. Pe de alta parte, din gena mutanta rezulta un numar mai mare de reziduuri de glutamina in comparatie cu gena normala (vezi figura 18.1.5.4), cauza fiind probabil o mutatie de tip „gain of function”; aceasta inseamna ca proteina sintetizata este functionala, dar are insa in plus efecte „toxice” datorita unei alterari a proprietatilor structurale si biochimice1;6. Date recente sugereaza ca interferarea lantului poliglutamic alungit cu transcriptia CBP (CREB binding protein), un mediator esential al semnalelor de supravietuire in neuronii maturi, ar constitui o “functie” dobandita genetic2.

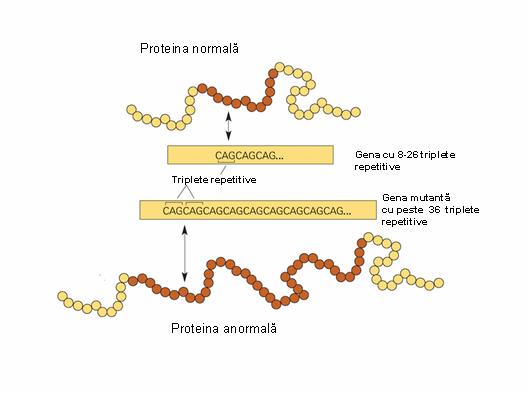
Fig. 18.1.5.4 Gena HTT normala si mutanta impreuna cu proteinele asociate
(Adaptare dupa publications.nigms.nih.govfindingssept08imageshunt_gene_big.jpg)
Huntingtina sufera modificari posttranslationale care constau in clivarea acesteia in aval de lantul poliglutamic (de catre enzimele denumite caspaze); fragmentul aminoterminal rezultat este citotoxic si provoaca apoptoza2;3. Acumularea intracelulara de fragmente aminoterminale de huntingtina agregate (incluzii intraneuronale) reprezinta una din caracteristicile neuropatologice ale coreei Huntington6.
Recomandari pentru testarea genetica
-confirmarea diagnosticului de coree Huntington la persoanele afectate;
-testarea in scop predictiv a adultilor asimptomatici cu risc crescut (50%); necesita o consiliere foarte atenta pre- si post-testare, deoarece nu este disponibil un tratament specific al bolii;
-diagnosticul prenatal la gravidele cu risc crescut (50%); se efectueaza prin analiza ADN-ul extras din celule fetale obtinute prin amniocenteza, de obicei efectuata la aproximativ 15-18 saptamani de gestatie, sau biopsia vilozitatilor coriale, la aproximativ 10-12 saptamani de gestatie; diagnosticul trebuie sa fie confirmat in familie prin tehnici moleculare, inainte de testarea prenatala5.
Specimen recoltat – sange venos4.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant4.
Cantitate recoltata – 5 mL sange4.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate proba – 7 zile la 2-8ºC4.
Metoda – reactie de polimerizare in lant (PCR) pentru detectarea tintita a alelor cu pana la 115 repetitii CAG4.
Raportarea si interpretarea rezultatelor
Va fi comunicat numarul de repetitii CAG in gena HTT4.
Alelele care contin ≥ 36 repetitii CAG sunt considerante ca fiind cauzatoare de boala si confera riscul de a dezvolta boala. In timp ce alelele cu 36-39 repetitii CAG au penetranta incompleta, cele cu ≥ 40 repetitii CAG sunt asociate intotdeauna cu dezvoltarea bolii.
Corelatii genotip-fenotip
Exista o corelatie inversa semnificativa intre numarul de repetitii CAG si varsta de debut a coreei Huntington:
• persoanele cu debut la varsta adulta prezinta de obicei un numar de repetitii CAG cuprins intre 36 si 55;
• persoanele cu debut juvenil prezinta de obicei un numar de repetitii CAG ≥60.
Mai multe informatii cu privire la varsta de debut in functie numarul de repetitii CAG se pot gasi pe website-ul: www.cmmt.ubc.ca.
De asemenea exista o corelatie negativa semnificativa intre dimensiunea segmentului de repetitii CAG si variabilitatea debutului, in sensul ca asocierea dintre dimensiunile CAG mai mici si debutul tardiv al bolii este mai putin precisa. In medie, marimea repetitiilor CAG este responsabila de pana la 70% din variabilitatea varstei de debut, in timp ce alti factori mosteniti sunt responsabili de 10-20% din variabilitatea reziduala.
Rata deteriorarii motorii, cognitive si a indicilor functionali se coreleaza direct cu marimea repetitiilor CAG. Se pare insa ca progresia tulburarilor comportamentale nu este asociata cu numarul repetitiilor CAG.
Homozigotii pentru alelele HTT cu penetranta completa au o varsta de debut a bolii similara cu cea a heterozigotilor, dar pot prezenta o rata accelerata de progresie a bolii.
Consilierea genetica
Coreea Huntington este o boala cu transmitere autozomal dominanta; din acest motiv copiii unei persoane afectate au un risc de 50% de a mosteni alela cauzatoare de boala. Daca persoana afectata este homozigota pentru expansiunea repetitiilor CAG, fiecare copil al sau va mosteni alela cauzatoare de boala.
La fratii persoanei afectate riscul de a face boala depinde de statusul genetic al parintilor:
• daca un parinte este afectat sau are o alela HTT cu ≥ 40 repetitii CAG riscul este de 50%;
• daca tatal are o alela HTT intemediara riscul de a mosteni o alela cauzatoare de boala este de pana la 5%; cu cat segmentul de repetitii CAG este mai mare cu atat este mai predispus la expansiune; nu a fost descris nici un caz de expansiune a unei alele intermediare materne; alelele intermediare expansionate sunt transmise preferential de barbatii cu varsta paterna avansata;
• un frate care mosteneste o alela HTT cu penetranta scazuta poate sau nu sa faca boala.
Desi majoritatea persoanelor afectate de coree Huntington au un parinte afectat, exista unele situatii in care istoricul familial este negativ:
• afectunea nu a fost diagnosticata corect la membri familiei;
• deces prematur al persoanei cu alela mutanta, inainte de debutul simptomelor;
• prezenta unei alele intermediare sau a unei alele cu penetranta redusa la un parinte asimptomatic;
• debut tardiv al bolii la parintele afectat.
Testarea genetica este recomandata la parintii unei persoane care prezinta o mutatie aparent de novo6.
Limite si interferente
In cazuri foarte rare pot fi depistate persoane varstnice asimptomatice avand un numar de 36-39 repetitii CAG.
Desi exista o corelatie importanta intre varsta de debut a bolii si marimea repetitiilor, CAG testarea genetica a persoanelor asimptomatice poate sa nu ofere intotdeauna o estimare adecvata a varstei de debut, a severitatii simptomelor sau a ratei de progresie a acestei afectiuni6.
Bibliografie
1. Francis O Walker. Huntington’s Disease. In The Lancet, 2007; 369:218-28.
2. Fredy J Revilla, Jaime Grutzendler, Travis R Larsh. Huntington Disease. www.emedicine.medscape.com, Ref Type: Internet Communication.
3. HTT Gene. Gene cards. www.genecards.org. Reference Type: Internet Communication.
4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
5. Rolf Knippers. Chorea Huntington. In Molekulare Genetik, 9. Auflage 2006, amazon.de.
6. Simon C Warby, Rona K Graham, Michael R Hayden. Huntington Disease. Gene Reviews, 2010. www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.
